I wrote this post whilst musing about the differences between frontier labs working on language models and biotech companies working on protein models, and specifically how biotech companies can approach the data bottleneck. I think the most valuable/successful biotech modelling platform companies will emerge in lead optimization rather than target discovery - but these thoughts are still very much a work in progress! Interested to hear alternative opinions/perspectives.
Binder design is hot right now. The field has quickly moved from methods like RFDiffusion and RFAntibody achieving a ~1% hit rate for finding <1µM binders to gradient-based inverse design methods (EvoBind2, BindCraft) that achieve binding rates of 10-100% across a wide range of target protein families. (good overviews from Boolean Biotech and Escalante Bio go into more detail for some of these methods). The development of these methods is very exciting from a scientific perspective! As designing binders becomes increasingly easy computationally, their range of use also expands significantly - for example, using de-novo proteins as pseudo-consumables in lab settings, for sensing or one-off assays.
The elephant in the room for these companies is access to differentiated experimental data. Most protein structure prediction models are trained on the PDB (or augmented versions thereof), the close to singular source of truth for experimentally solved protein structures. While there is also a massive body of private data for training text/vision LLMs (Youtube being the most obvious example), the data is not categorically different from existing data available on the web. This is not the case with biological data - Big pharma has access to vast amounts of proprietary data, knows this data is valuable, and isn't going to give it away for free.
This data bottleneck has two, equally ferocious heads. Firstly, the available data is quite small - the PDB contains 229,564 structures, but when clustered by sequence similarity, the size of datasets used for training is much smaller - typically of the order of 10,000-40,000 structures. Secondly, the PDB is overwhelmingly dominated by naturally occurring proteins, with only a small fraction of the structures being synthetic or engineered proteins, and biases towards certain protein families and tertiary structures. The Proteina paper (Table 4) demonstrates some of the potential issues that this data bottleneck can create by showing that unconditional folding models generate structures with predominantly alpha-helices over 70% of the time.
In addition to this data bottleneck, the additional domain advantage for working in the natural sciences is rapidly falling, with companies like Adaptyv offering white-glove experimental design services, and models like SimpleFold showing that in the limit of more data, the complex, domain-specific algorithmic designs of some structure prediction modules are not necessary. There is likely some open road ahead in the form of smart approaches to data augmentation and pretraining, particularly using pLDDT based uncertainty metrics, but fundamentally, there is a data bottleneck which will remain impassable without direct experimental feedback loops.
It is notable that the most promising companies operating in the applied biology space have distinct theses for how to resolve these issues with a platform-based technology (A-Alpha Bio with multiplexed protein-protein interaction assays, Manifold Bio with in-vivo screening, Nabla Bio with a therapeutically relevant wet lab setup for antibodies).
For a foundational research company to succeed at tackling the more general version of this problem, they need to be able to generate models capable of exploring the vast, unnatural sequence space of protein sequences, despite models that are trained only on protein databases drawn broadly from the evolutionary distribution of proteins. Given this computational model <> experimental structure gap, two questions naturally follow.
How do we most efficiently bridge this gap from a research and engineering perspective?
Which businesses/business models are most well placed to source the necessary data, information and talent to be able to execute on this objective? (for a followup post)
This post will focus on an approach that I think is quite promising for addressing question 1 - high throughput mutagenesis, or deep mutational scanning (DMS).
What is Deep Mutational Scanning?
Deep mutational scanning (DMS) is a technique for exploring the functional landscape of proteins by systematically introducing mutations across the protein sequence and measuring the resulting changes in protein function. A typical DMS experiment involves three steps: 1) generating a genetic mutant library; 2) performing a high-throughput phenotyping assay; 3) and deep sequencing and data analysis to identify the functional impact of each mutation. Mutagenesis is used across discovery and lead optimization to both identify and validate potential epitopes (places on the protein that are important for function), as well as a lead optimization technique for improving protein function/stability/expression.
There are 3 main types of mutagenesis: Alanine scanning, site saturation mutagenesis and random mutagenesis.
Alanine scanning
Alanine scanning involves creating mutant libraries by substituting every position in a target protein with the amino acid alanine. It is commonly used for “hotspot detection” or epitope mapping - identifying sub-sequences or motifs that are critical for protein function.
Alanine, rather than other amino acids, is chosen because it has a small methyl group that doesn't introduce extreme steric or electrostatic effects. This means that proteins with residues substituted for Alanine typically retain the same secondary structure - meaning their overall 3D structure is generally conserved. This property of Alanine is the basis for the wonderfully named “Alanine World Model”, which postulates that Alanine is the base amino acid from which polar amino acids (GCA coded) and non-polar GCAU are derived, forming the secondary structure motifs of α-helices and β-sheets respectively.
Even this relatively simple technique can allow quite advanced insight into protein function. The original paper introducing Alanine Scanning used it to identify the critical residues in the receptor binding domains for Human Growth Hormone (hGH). During the pandemic, Naveenchandra Suryadevara and co-workers at the Vanderbilt Vaccine Center used alanine scanning to map hotspots of the N-terminal domain of SARS-CoV-2. Most diagnostic tests and available vaccines for COVID-19 were designed based on the larger “Spike” protein - where the virus attaches to healthy cells (the receptor binding domain). Vaccines targeting this spike domain work well, but suffer from one critical flaw - receptor binding domain in many viruses is highly variable, meaning that new variants of the virus are likely to have mutations in this region, reducing the effectiveness and shelf-life of vaccines. In contrast, the N-terminal domain is highly conserved - so antibodies targeting this protein region may be more effective across virus variants.
Site saturation mutagenesis
In contrast to Alanine scanning which replaces each different position in a protein sequence with the same (alanine) residue, site saturation mutagenesis replaces a single position in a protein sequence with every possible alternative residue. This type of mutagenesis is a common protein engineering tool across antibody affinity maturation, enzyme engineering and binder design, given the requirement for knowing which residues are important upfront.
Random mutagenesis
Random mutagenesis differs from the other two methods in that it does not use a deterministic mutagenesis protocol, but rather a stochastic one. Mutations are introduced randomly to the protein sequence, and the resulting variants are then tested for their functional impact. This is often achieved using error-prone PCR, where the DNA polymerase is mutagenic and introduces random mutations to the DNA sequence. This method for generating random mutagenesis libraries is relatively cheap and easy to scale.
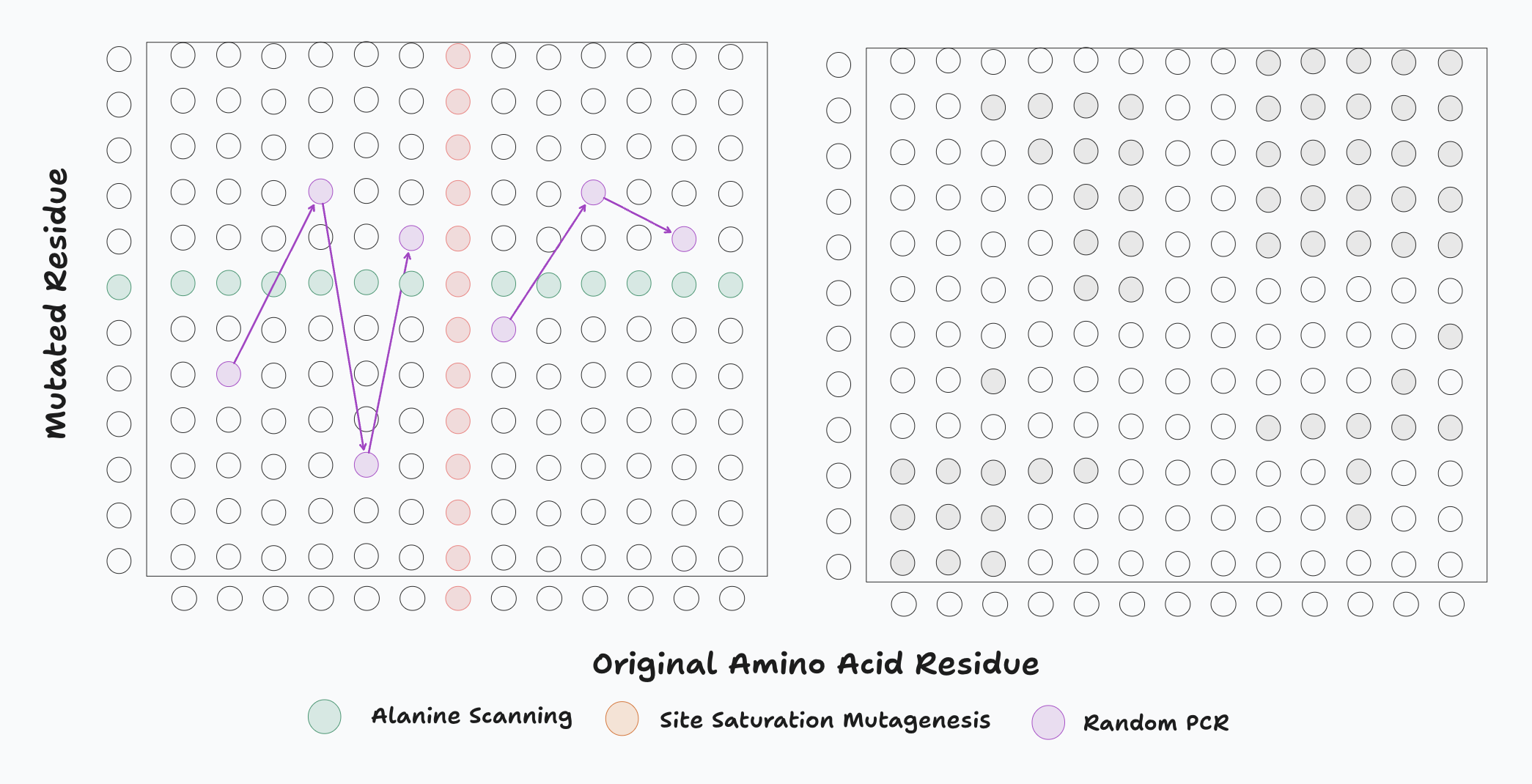
Left: DNA libraries generated by random mutagenesis in sample sequence space. Each set of connected dots/coloured dots represents one approach to mutagenesis. Error-prone PCR randomly mutates some residues to other amino acids. Alanine scanning replaces each residue of the protein with alanine, one-by-one. Site saturation substitutes each of the 20 possible amino acids (or some subset of them) at a single position, one-by-one. Right: The ideal experimental exploration of sequence space, based on 1) a constrained search space and 2) driven by computational model likelihood for given mutations within the constrained regions.
Once libraries have been generated and expressed into the cell type of interest, the resulting variants are sequenced using next-generation sequencing (NGS). The assays typically fall into two categories - fitness assays and functional assays. Fitness assays measure the relative fitness of the variants compared to the wild type based on growth rates/expression levels only. These are cheap and easy to run, but do not necessarily provide insight into the functional aspect of each mutation - just whether or not it is beneficial for expression. Functional assays measure the impact of each mutation on protein function. These are more expensive and time-consuming as they are typically specific to the function of interest and require more complex assay conditions (reporters, subsequent readouts using fluorescent or spectroscopic assays), but provide more insight into the functional aspect of each mutation.
Each of these produce fitness scores/counts compared to the wild type, which can be used to generate a relative quality score for each mutation. Tools for manipulating these datasets, such as enrich_2 and DiMSum, can be used to generate a relative quality score for each mutation. A good summary of these tools and why they are necessary is available in this review article.
Mutagenesis techniques generate data which is particularly suitable for alignment of protein language models. It gives a relative metric of quality between a wild type (original) structure and a mutated structure. If one squints their eyes (quite hard), this data looks similar to the human preference data that is commonly used to align text based language models - we have multiple generations, with a relative quality score describing an implicit preference. In fact, mutagenesis data is already used widely for evaluation of protein representation learning - the Protein Gym benchmark being the primary example.
Mutagenesis data overcomes the primary problem with the existing datasets available to any protein modelling method - all of the experimentally derived structures are from the evolutionary distribution of proteins. Capturing relative quality measures of proteins near, but not on this data manifold is one possible strategy for beginning to explore the space of unnatural protein sequences.
Modelling using these ideas
Although mutagenesis data has broadly been used for evaluating protein representation learning techniques, several recent papers have demonstrated that this alignment approach using DMS data can work well experimentally. There are quite a few promising papers in this space, but I have picked out two that I think are particularly interesting/well written.
Functional alignment of protein language models via reinforcement learning introduces a method called “Reinforcement Learning from Experiment Feedback (RLXF)” (essentially RLHF but for mutagenesis data) to engineer CreiLOV, an oxygen-independent fluorescent reporter protein. CreiLOV is interesting because the ubiquitous oxygen-dependent green fluorescent protein (GFP) is oxygen-dependent, meaning it cannot be used in hypoxic or anaerobic environments such as gut microbiomes or high-density fermentations. CreiLOV is also a great example of why evolutionary preferences in a protein may not be functionally aligned.
Proteins containing the LOV domain (of which CreiLOV is one) typically occur as blue light photoreceptors in fungi, plants and algae. As such, fluorescence is not required for their function - it’s just a happy accident of evolution. The authors demonstrate this by showing that functionally improved CreiLOV mutants are not preferred by protein language models, highlighting the need for this type of functional alignment.
Accelerating protein engineering with fitness landscape modelling and reinforcement learning (code available) from Microsoft Research takes a similar line, adding in a search component. The particularly exciting thing about this paper is that they show generalization from models trained on single mutation data to generations containing multiple mutants. This is interesting - one downside of mutagenesis data is that it doesn’t capture epistasis (the functional co-dependence between individual mutations made to a protein sequence) as in its most common forms, it only generates single mutation variants. This suggests that even with standard mutagenesis data, we might be able to align PLMs.
Why Shouldn’t we use Mutagenesis data?
Using mutagenesis data to train or fine-tune models is far from straightforward. Experimental readouts of “function” depend heavily on the cell type, assay conditions, and post-translational modifications which all shape what’s actually being measured. The same mutation in one protein can enhance binding in one environment and diminish expression in another, making it difficult to define a stable reward signal across different domains/cell types. Some assay readouts, like expression or thermostability, do generalize across hosts/domains, but most don’t. Finding generalizable assays which provide value across a wide range of targets and conditions is a major challenge.
Typical DMS datasets are single mutation variants, which is not sufficient to capture the epistatic interactions that drive true functional shifts. Indels (deletions and insertions) are also not captured by single mutation data, and are a major source of functional variation in proteins. For alignment, we need richer objectives - either through multi-mutation experimental datasets or by integrating mutational feedback with large-scale textual or structural data that encode biological context. Some data in this form already exists - for example the Disease and Variants tab of UniProt contains known mutations and their effects on protein function, along with citations to the papers demonstrating these effects.
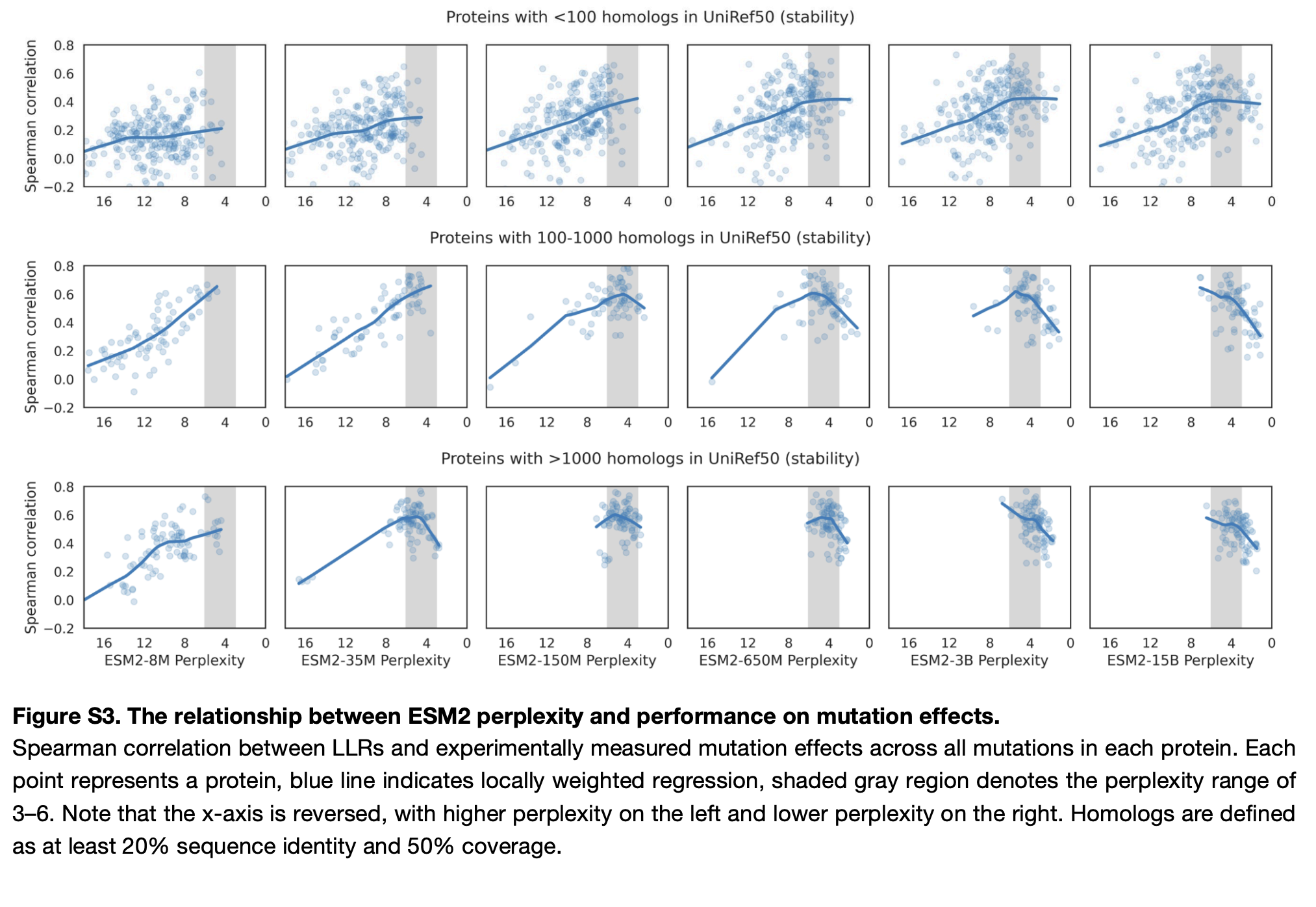
The ability to evaluate zero-shot performance of variant prediction models on single mutation data is currently a major point of debate in the community. This paper from Deborah Mark's lab find that simple alignment approaches are competitive with parameterized methods for viral proteins (out of distribution compared to ProteinGym). Pascal Notin, the lead author of ProteinGym, also has a sobering blog post on scaling laws for biology, although some of these observations have been explained by Hou Chao's excellent work showing a correlation between model perplexity, MSA depth/sequence similarity and Spearman correlation for some of the ProteinGym and Stability DMS datasets.
Other tailwinds
In addition to the existing promising RL approaches being applied to mutagenesis data, there are a few other tailwinds that make pursuing this space quite attractive.
Firstly, the cost of running mutagenesis experiments is falling rapidly. Traditional DNA synthesis has long faced a trade-off between sequence length and scale. Biologists could either synthesize a few long fragments (<5,000 bp) or many short oligopools (~300 bp). Companies like Twist Bioscience and IDT are now extending these limits with multiplexed gene fragments up to 500 bp, allowing the construction of large libraries for mutational scanning. New techniques for creating libraries combinatorially from oligopools massively reduce the cost and complexity of designing and running these experiments at scale. Combined with ML based optimization methods for selecting library candidates, this could reduce the cost of running mutagenesis experiments to a fraction of what it is today.
Secondly, mutational scanning does not only apply to protein engineering campaigns. It is already a widely used technique for understanding genetic variation, with MAVE (Multiplexed Assays of Variant Effects) and MAVE DB (a database of MAVE assays) being established tools for genetics. MAVE assays are a type of mutational scanning assay that is used to identify the functional impact of genetic variants, typically through measuring disease co-occurence or changes in drug response. Integrating these assays with ML based approaches to variant effect prediction could provide a rich source of data for multimodal proteomic/genomic language model alignment whilst also providing immediate value in the form of predictive risk screening, clinical trial selection and target identification for precision medicine. New methods like LABEL-seq for in-situ, multiplexed measurement of variants would push this even further.
Finally, mutagenesis data has typically been constrained to a fixed number of mutations. This makes a lot of sense when trying to make scientific interventions, as it allows us as humans to generate hypotheses for the causal mechanisms underlying protein function. However, pLMs offer us a method to explore this sequence space in a much more efficient manner. Single and low-N mutant datasets constrain our ability to explore this sequence space with models - but there is no reason we can't use existing random mutagenesis techniques to generate much larger datasets with unconstrained numbers of residues mutated. This process could even be driven by a model - directly pointing to an exploration/exploitation trade-off for exploring sequence space.
Mutagenesis data offers a potential bridge between experimental biology and machine learning by offering a way to connect models trained on the evolutionary distribution of natural proteins to the far larger space of synthetic proteins. Deep mutational scanning has several attractive attributes - it is a well-understood technique with a long history of use in protein engineering; it provides relative quality measures that resemble preference data used in LLM alignment; and experimental costs are falling rapidly due to advances in DNA synthesis and combinatorial library construction.
If you enjoyed this post or want to chat about anything in it, please feel free to reach out! I always enjoy talking about these types of technical problems.